Xiao-Dong Yang ![]() ,
Wan-Sen Li,
Yan-Juan Tian,
Cheng-Gong Liu,
Da-Hong Gao,
Hai-Li Ma
,
Wan-Sen Li,
Yan-Juan Tian,
Cheng-Gong Liu,
Da-Hong Gao,
Hai-Li Ma
For correspondence:- Xiao-Dong Yang Email: yangxd8891@gmail.com Tel:+863962926209
Received: 22 July 2015 Accepted: 5 May 2016 Published: 28 June 2016
Citation: Yang X, Li W, Tian Y, Liu C, Gao D, Ma H. Dissolution rate enhancement of repaglinide by solid dispersion. Trop J Pharm Res 2016; 15(6):1123-1128 doi: 10.4314/tjpr.v15i6.2
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To enhance the solubility and dissolution rate of the antidiabetic drug repaglinide by solid dispersion (SD) technique
Method: The solid dispersion of repaglinide was prepared by solvent evaporation method using the hydrophilic carrier, polyethylene glycol 4000 (PEG 4000) in three drug:PEG 4000 ratios (1:1, 1:3, 1:5). For comparison, physical mixtures of repaglinide and PEG 4000 in the same ratios were also prepared. The formulations were characterized by Fourier transformed infrared spectroscopy (FTIR), x-ray diffractometry (XRD) and differential scanning colorimetry (DSC). Phase solubility study of pure repaglinide, physical mixture and solid dispersion was performed in distilled water. Dissolution studies were carried out in pH 7.4 phosphate buffer.
Results: DSC and XRD results indicate that repaglinide exists in amorphous form in solid dispersion. FT-IR analysis demonstrated the presence of intermolecular hydrogen bonding between repaglinide and PEG 4000 in the solid dispersion. The solubility of pure repaglinide was enhanced from 22.5± 5.0 to 235.5± 5.0 µg/mL in distilled water at 37 0C. Rapid burst release (80 - 86 %) from the solid dispersion formulations was observed within 15 min.
Conclusion: The solubility and dissolution rate of repaglinide are enhanced by formulating SDs of repaglinide with PEG 4000. This will likely lead to increase in bioavailability which would be beneficial for better glucose control in diabetic patients.
Introduction
The oral bioavailability of a pharmaceutically active ingredient depends on its water solubility and dissolution rate. Rate of dissolution is the rate determining step for the onset of therapeutic activity [1].Water insoluble drugs are usually characterized by a low bioavailability due to less absorption which is a major concern of pharmaceutical industries worldwide [2].Micronization, co-crystal formation, cyclodextrin complexation, salt formation, use of surface active agents, co-solvency are some of the approaches to improve the dissolution rate of the drugs [3]. Solid dispersion (SD) is one of the most widely used techniques to improve solubility as well as dissolution rate of poorly water soluble drugs. This method involves a dispersion of one or more active ingredients in an inner carrier or matrix in solid state prepared by melting, dissolution in solvent or melting solvent method [4].
SD is generally prepared by solvent method or fusion method. In solvent method, the drug and carrier are dissolved in a mutual organic solvent followed by solvent evaporation, whereas in fusion method, drug-carrier mixtures are prepared by co-melting and cooling. In both methods, a suitable carrier needs to be selected in order to achieve enhanced solubility [5]. Hydrophilic synthetic polymers have been widely investigated as carrier substances for solid dispersions. Polyethylene glycol (PEG) and polyvinylpyrrolidone (PVP) are amongst the most frequently investigated hydrophilic polymeric carriers. SD using suitable carrier, changes the micro-environment of the drug particles, reduces the drug particle size thereby increasing dissolution, solubility and thus changes biopharmaceutical properties [6].
Repaglinide is an antidiabetic drug in the class of medications known as meglitinides, and was invented in 1983. Repaglinide lowers blood glucose by stimulating the release of insulin from the pancreas. It achieves this by closing ATP-dependent potassium channels in the membrane of the beta cells. This depolarizes the beta cells, opening the cells' calcium channels, and the resulting calcium influx induces insulin secretion. Repaglinide is a poorly water-soluble compound, with water solubility of approximately 20 μg/mL, high lipophilicity (logP = 3.97) and relatively low oral bioavailability (56 %) which is attributed to poor dissolution (BCS class II) and it undergoes hepatic first pass metabolism [7-9].
The aim of the present study was to prepare and characterize solid dispersion of repaglinide so as to improve its dissolution property and bioavailability. Solubility and dissolution tests were carried out to evaluate the effectiveness of the carriers for repaglinide.
Methods
Repaglinide was received as a gift from Zhejiang Haixiang Pharmaceutical Co., Ltd. (Zhejiang, China) while PEG 4000 was received from Clariant (Germany). Double distilled water was used throughout the study and all the other chemicals used were of analytical grade.
Preparation of SD by solvent evaporation method
SD of RPG was prepared in PEG 4000 by solvent evaporation method [10]. For solvent evaporation method, RPG was dissolved in sufficient quantity of acetone; to this solution an appropriate amount of PEG 4000 was added. The SDs of RPG in PEG 4000 in three different weight ratios (1:1, 1:3, 1:5) were prepared. The solvent was evaporated under reduced pressure at 45 °C by using rotary evaporator and the resulting residue dried under vacuum for 4 h. The mixture was stored overnight in a desiccator. The hardened mixture was powdered in a mortar, sieved through a 100- mesh screen (149 µ), and stored in a screw-cap vial at room temperature until further use.
Physical mixtures were prepared by mixing RPG with PEG 4000 at the same weight ratios (1:1, 1:3, 1:5) in a mortar until a homogeneous mixture was obtained. The physical mixtures were passed through a 100 mess sieve and stored in a screw-cap vial at room temperature until further use.
Phase solubility study
Excess amount of pure RPG, solid dispersion containing RPG and physical mixtures was added to 5 mL of distilled water in screw cap vials to ensure the drug reached saturation level. These solutions were mechanically shaken for 24 h at 37 oC and then the solutions were centrifuged at 15,000 rpm for 3 min [11]. The saturated solutions were then diluted to proper concentration and absorbance was measured at 243.5 nm and solubility in each solution was determined.
Dissolution studies
The SD and physical mixture equivalent to 15 mg of RPG were weighed and added into the dissolution medium (pH 7.4 phosphate buffer, 900 ml) maintained at 37 ± 0.5 oC. At the specified times 5 mL samples were withdrawn by using syringe filter and then assayed for RPG content by measuring the absorbance at 247.5 nm using a UV-Visible spectrophotometer (UV-1800, Shimadzu, Japan). Dissolution studies were performed in triplicate (n = 3), and mean values taken.
FT-IR study
FT-IR spectra were obtained by an FT-IR spectrophotometer (Bruker, Germany) using the potassium bromide (KBr) disk method. Data were collected over a spectral region from 4000 to 400 cm-1 with a resolution of 2 cm-1.
Differential scanning calorimetry (DSC)
The DSC measurements were performed on a DSC-6100 differential scanning calorimeter with a thermal analyzer (Seiko Instruments, Japan). All accurately weighed samples were placed in sealed aluminum pans, before heating under nitrogen flow (20 mL/min) at a scanning rate of 10 °C min−1 from 25 to 250 °C. An empty aluminum pan was used as reference.
XRD measurement
XRD measurement was carried out using a powder x-ray diffractometer (Thermo Scientific, Waltham, Massachusetts, USA) with Ni-filtered Cu Kα radiation, a tube voltage of 40 kV and a tube current of 40 mA. The scanning rate was 10 °/min over a 2θ range of 5 – 40° with a step size of 0.02°.
Results
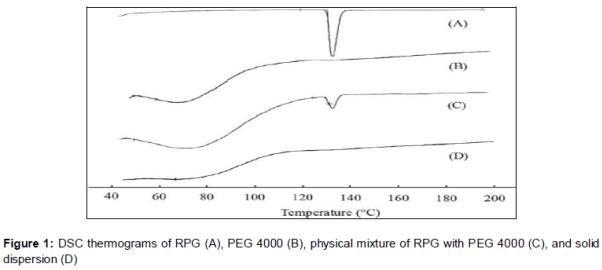
DSC thermograms of RPG, PEG 4000, physical mixture of RPG with PEG 4000, and solid dispersion are shown in . Pure RPG and its physical mixture with PEG 4000 exhibited an endothermic peak at around 135 °C, which was consistent with the melting point of repaglinide. The small peak in physical mixture might be explained by the dilution effect of PEG 4000. However, no such peaks were observed in the PEG 4000 and solid dispersion group. The former resulted from the amorphous nature of the polymer, and the later revealed no crystallinity of repaglinide existed in solid dispersion, indicating that repaglinide might be in an amorphous form.
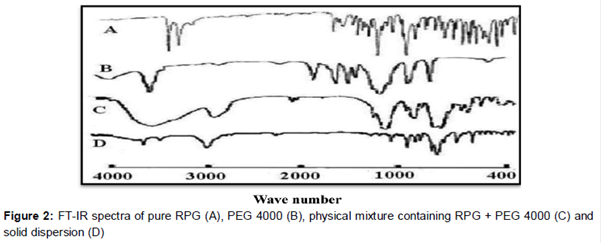
shows the FT-IR spectra of pure RPG (A), PEG 4000 (B), physical mixture containing RPG + PEG 4000 (C) and solid dispersion (D). Characteristic absorption bands of pure RPG (A) were observed at 3310, 1570, 1085, 915 and 783 cm−1. These absorption bands also existed at the FT-IR spectrum of physical mixture (C), revealing that there was no interaction between RPG and PEG 4000 in the physical mixture. In contrast, the absorption band attributed to O–H stretching vibration of repaglinide disappeared in the FT-IR spectrum of solid dispersion (D). It might be the result of the formation of hydrogen bonding between repaglinide and PEG 4000.
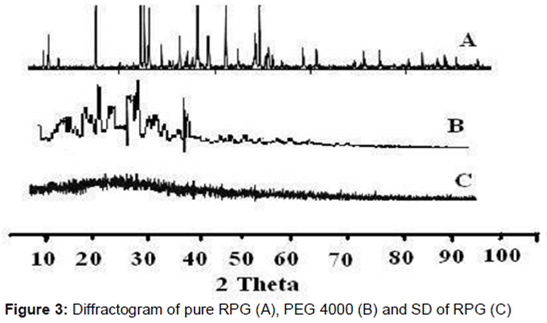
shows the diffractogram of pure RPG (A), PEG 4000 (B) and SD of RPG (C). RPG showed the characteristic intense peaks at 2θ of 20, 30, 33, 44 and 56.50. These peaks clearly indicated crystalline nature of the pure RPG. Also, the characteristic intense peaks for PEG 4000 were observed at 2θ of 22.5, 29 and 37.50. The intensity of these peaks from RPG and PEG 4000 disappeared in diffractogram of the SD as observed in C. This pattern indicates the amorphous nature of RPG in SD.
Phase solubility
RPG belongs to the BCS class II drug, i.e., low soluble and high permeable in nature. As discussed before it is a poorly water soluble drug having log p value of 3.97.The intrinsic aqueous solubility of pure RPG was determined and it was found to be to be 22.5 ± 5 µg/mL. This intrinsic solubility of the pure RPG was found to be equivalent to reported solubility in previously published literature [12].
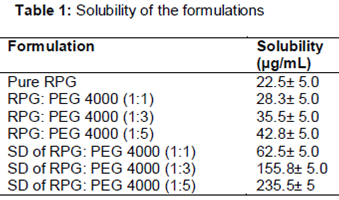
So to enhance the aqueous solubility of RPG, physical mixture with hydrophilic carrier PEG 4000 and SD were prepared. It can be found that the solubility was enhanced in all physical mixtures and SD’s. The solubility of RPG was increased by increasing the concentration of the hydrophilic carrier in both physical mixture and SD. In case of physical mixture, maximum solubility of 42.8 ± 5 was found. Very promising results were obtained in case of the SD. The solubility was enhanced from 22.5 ± 5 to 235.5 ± 5 µg/mL, i.e., ~ 10 times solubility was enhanced when RPG was converted to the SD. The solubility data are presented in .
In vitro dissolution
Dissolution study of pure RPG, physical mixtures and SD was performed in pH 7.4 phosphate buffer. shows the dissolution profiles of repaglinide, its physical mixture and solid dispersion with PEG 4000. These three formulations exhibited wide difference in dissolution pattern. The fastest one was solid dispersion, followed by the physical mixture followed by pure RPG. The rapid burst release (80 -86 %) was observed within 15 min in case of SD formulations. It clearly indicated the ideal complexation between the RPG and PEG 4000. The possible reason for the enhancement of the drug release from SD was the reduction in crystalline nature of pure of RPG and conversion in to the more soluble amorphous nature. The reduction in particle size as well as enhancement of the wettability of the drug may also be the probable reason for the enhancement of the drug release. The drug release pattern of the physical mixture found to be the lower as compare to the SD. This was due to the existence of the drug in crystalline form that led to more energy to break up the crystal structure. So it was clear that the SD technique was vital tool for the enhancement of the solubility as well as the dissolution profile of poorly water soluble drug [13].
Discussion
Increased wettability, alteration of surface properties of drug particle, formation of amorphous complex, solubilization effect of PEG 4000, reduction of particle aggregation of the drug, might be responsible for the enhanced solubility and dissolution rate of RPGL from its SD and to some extent in PMs. The solubility of RPG was increased by increasing the concentration of the hydrophilic carrier in both physical mixture and SD. The fastest dissolution rate was found for solid dispersion, followed by the physical mixture followed by pure RPG. The rapid burst release (80-86 %) was observed within 15 min in case of SD formulations. From FTIR spectroscopy, it was concluded that there was no well-defined chemical interaction between RPGL and PEG 4000 in SDs and in physical mixture. No endothermic peak of RPGL was present in the DSC thermograms of SDs with PEG 4000 suggesting the absence of crystalline RPGL, however, the XRD studies indicated the complete amorphous nature of the RPG in SDs.
Conclusion
The solubility and dissolution rate of RPG can be enhanced by formulating SDs of RPG with PEG 4000. Enhancement of solubility and dissolution rate will likely increase bioavailability which would be beneficial for better glucose control in diabetic patients. However, in vivo studies are required to ascertain this.
Declarations
Acknowledgement
References
Archives


News Updates